Image: peritoneal endometriotic lesions expressing Fra2
PIK3CA mutation in endometriotic epithelial cells promotes viperin-dependent inflammatory response to insulin (2023, Reproductive Biology and Endocrinology)
Mike Wilson, Shannon Harkins, Jake J. Reske, Rebecca A. Siwicki, Marie Adams, Victoria Bae-Jump, Jose Teixeira, & Ronald L. Chandler.
PMID: 37170094. Journal link.
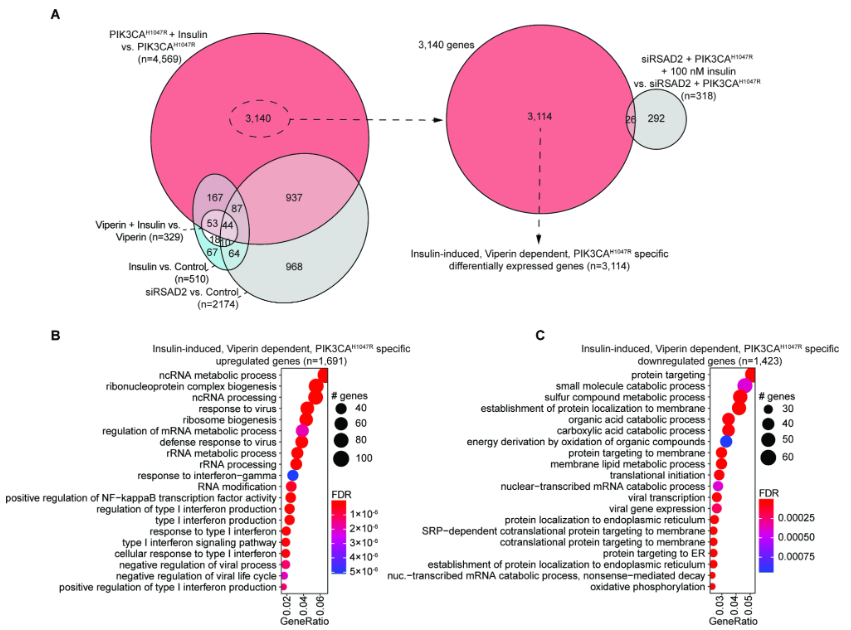
Endometrial epithelia are known to harbor cancer driver mutations in the absence of any pathologies, including mutations in PIK3CA. Insulin plays an important role in regulating uterine metabolism during pregnancy, and hyperinsulinemia is associated with conditions impacting fertility. Hyperinsulinemia also promotes cancer, but the direct action of insulin on mutated endometrial epithelial cells is unknown. Here, we treated 12Z endometriotic epithelial cells carrying the PIK3CAH1047R oncogene with insulin and examined transcriptomes by RNA-seq. While cells naively responded to insulin, the magnitude of differential gene expression (DGE) was nine times greater in PIK3CAH1047R cells, representing a synergistic effect between insulin signaling and PIK3CAH1047R expression. Interferon signaling and the unfolded protein response (UPR) were enriched pathways among affected genes. Insulin treatment in wild-type cells activated normal endoplasmic reticulum stress (ERS) response programs, while PIK3CAH1047R cells activated programs necessary to avoid ERS-induced apoptosis. PIK3CAH1047R expression alone resulted in overexpression (OE) of Viperin (RSAD2), which is involved in viral response and upregulated in the endometrium during early pregnancy. The transcriptional changes induced by insulin in PIK3CAH1047R cells were rescued by knockdown of Viperin, while Viperin OE alone was insufficient to induce a DGE response to insulin, suggesting that Viperin is necessary but not sufficient for the synergistic effect of PIK3CAH1047R and insulin treatment. We identified interferon signaling, viral response, and protein targeting pathways that are induced by insulin but dependent on Viperin in PIK3CAH1047R mutant cells. These results suggest that response to insulin signaling is altered in mutated endometriotic epithelial cells.
PMID: 37170094. Journal link.
Endometrial epithelia are known to harbor cancer driver mutations in the absence of any pathologies, including mutations in PIK3CA. Insulin plays an important role in regulating uterine metabolism during pregnancy, and hyperinsulinemia is associated with conditions impacting fertility. Hyperinsulinemia also promotes cancer, but the direct action of insulin on mutated endometrial epithelial cells is unknown. Here, we treated 12Z endometriotic epithelial cells carrying the PIK3CAH1047R oncogene with insulin and examined transcriptomes by RNA-seq. While cells naively responded to insulin, the magnitude of differential gene expression (DGE) was nine times greater in PIK3CAH1047R cells, representing a synergistic effect between insulin signaling and PIK3CAH1047R expression. Interferon signaling and the unfolded protein response (UPR) were enriched pathways among affected genes. Insulin treatment in wild-type cells activated normal endoplasmic reticulum stress (ERS) response programs, while PIK3CAH1047R cells activated programs necessary to avoid ERS-induced apoptosis. PIK3CAH1047R expression alone resulted in overexpression (OE) of Viperin (RSAD2), which is involved in viral response and upregulated in the endometrium during early pregnancy. The transcriptional changes induced by insulin in PIK3CAH1047R cells were rescued by knockdown of Viperin, while Viperin OE alone was insufficient to induce a DGE response to insulin, suggesting that Viperin is necessary but not sufficient for the synergistic effect of PIK3CAH1047R and insulin treatment. We identified interferon signaling, viral response, and protein targeting pathways that are induced by insulin but dependent on Viperin in PIK3CAH1047R mutant cells. These results suggest that response to insulin signaling is altered in mutated endometriotic epithelial cells.
Obesity alters the mouse endometrial transcriptome in a cell context-dependent manner (2022, Reproductive Biology and Endocrinology)
Mike Wilson, Hilary Skalski, Jake J. Reske, Marc Wegener, Marie Adams, Galen Hostetter, Hanne M. Hoffmann, Jamie Bernard, Victoria Bae-Jump, Jose Teixeira, & Ronald L. Chandler.
PMID: 36424602. Journal link.
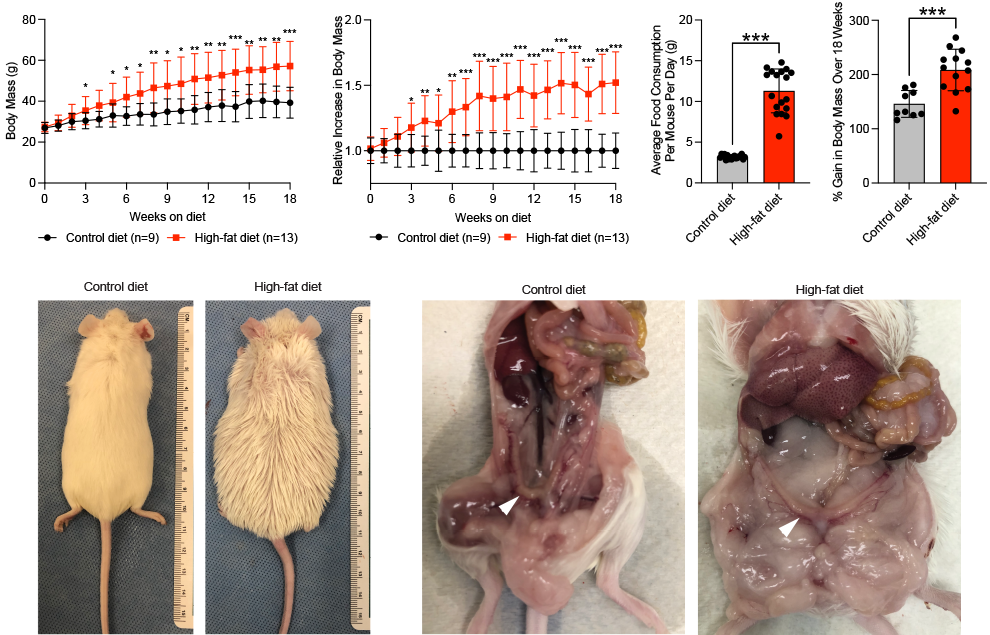
Obesity impacts fertility and is positively correlated with endometrial hyperplasia and endometrial cancer occurrence. Endometrial epithelia often harbor disease driver-mutations, while endometrial stroma are highly regulative of neighboring epithelia. Here, we sought to determine distinct transcriptome changes occurring in individual cell types in the obese mouse uterus. Outbred CD-1 mice were fed high-fat or control diets for 18 weeks, estrous cycle staged, and endometrial epithelia, macrophages, and stroma isolated for transcriptomic analysis. High-fat diet mice displayed increased body mass and developed glucose intolerance, hyperinsulinemia, and fatty liver. Obese mouse epithelia displayed differential gene expression for genes related to innate immunity and leukocyte chemotaxis. The obese mouse stroma differentially expressed factors related to circadian rhythm, and expression of these genes correlated with glucose tolerance or body mass. We observed correlations between F4/80 + macrophage numbers, Cleaved Caspase 3 (CC3) apoptosis marker staining and glucose intolerance among obese mice, including a subgroup of obese mice with high CC3 + luminal epithelia. This subgroup displayed differential gene expression among all cell types, with pathways related to immune escape in epithelia and macrophages, while the stroma dysregulated pathways related to regulation of epithelia. These results suggest an important role for differential response of both the epithelia and stroma in their response to obesity, while macrophages are dysregulated in the context of apoptotic epithelia. The obesity-related gene expression programs in cells within the uterine microenvironment may influence the ability of the endometrium to function during pregnancy and influence disease pathogenesis.
PMID: 36424602. Journal link.
Obesity impacts fertility and is positively correlated with endometrial hyperplasia and endometrial cancer occurrence. Endometrial epithelia often harbor disease driver-mutations, while endometrial stroma are highly regulative of neighboring epithelia. Here, we sought to determine distinct transcriptome changes occurring in individual cell types in the obese mouse uterus. Outbred CD-1 mice were fed high-fat or control diets for 18 weeks, estrous cycle staged, and endometrial epithelia, macrophages, and stroma isolated for transcriptomic analysis. High-fat diet mice displayed increased body mass and developed glucose intolerance, hyperinsulinemia, and fatty liver. Obese mouse epithelia displayed differential gene expression for genes related to innate immunity and leukocyte chemotaxis. The obese mouse stroma differentially expressed factors related to circadian rhythm, and expression of these genes correlated with glucose tolerance or body mass. We observed correlations between F4/80 + macrophage numbers, Cleaved Caspase 3 (CC3) apoptosis marker staining and glucose intolerance among obese mice, including a subgroup of obese mice with high CC3 + luminal epithelia. This subgroup displayed differential gene expression among all cell types, with pathways related to immune escape in epithelia and macrophages, while the stroma dysregulated pathways related to regulation of epithelia. These results suggest an important role for differential response of both the epithelia and stroma in their response to obesity, while macrophages are dysregulated in the context of apoptotic epithelia. The obesity-related gene expression programs in cells within the uterine microenvironment may influence the ability of the endometrium to function during pregnancy and influence disease pathogenesis.
ARID1A-dependent maintenance of H3.3 is required for repressive CHD4-ZMYND8 chromatin interactions at super-enhancers (2022, BMC Biology)
Jake J. Reske, Mike Wilson, Brooke Armistead, Shannon Harkins, Cristina Perez, Joel Hrit, Marie Adams, Scott B Rothbart, Stacey Missmer, Asgerally Fazleabas & Ronald L. Chandler.
PMID: 36153585. Journal link.
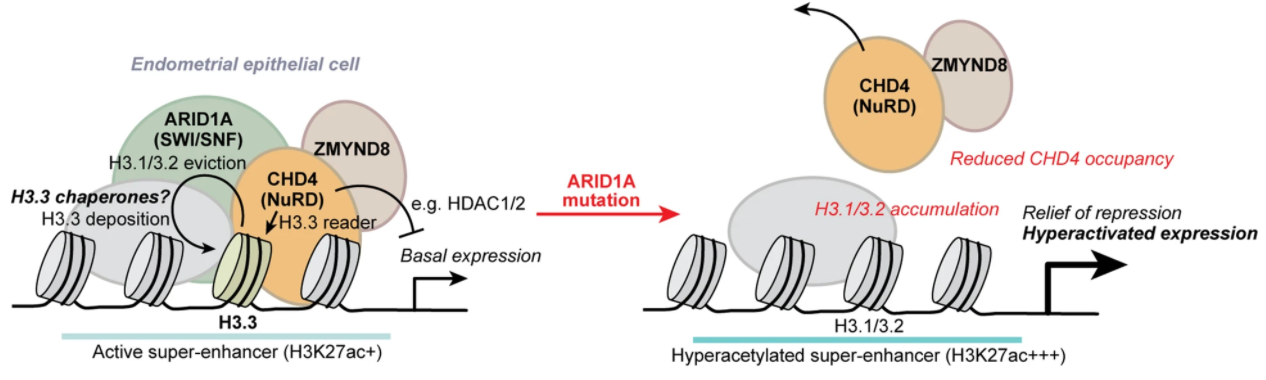
SWI/SNF (BAF) chromatin remodeling complexes regulate lineage-specific enhancer activity by promoting accessibility for diverse DNA-binding factors and chromatin regulators. Through genome-wide analysis of human endometriotic epithelial cells, we show that more than half of ARID1A binding sites are marked by the variant histone H3.3, including active regulatory elements such as super-enhancers. ARID1A knockdown leads to H3.3 depletion and gain of canonical H3.1/3.2 at ARID1A-bound active regulatory elements, and a concomitant redistribution of H3.3 toward genic elements. ARID1A interactions with the repressive chromatin remodeler CHD4 (NuRD) are associated with H3.3, and ARID1A is required for CHD4 recruitment to H3.3. ZMYND8 interacts with CHD4 to suppress a subset of ARID1A, CHD4, and ZMYND8 co-bound, H3.3+ H4K16ac+ super-enhancers near genes governing extracellular matrix, motility, adhesion, and epithelial-to-mesenchymal transition. Thus, ARID1A-containing BAF complexes are required for maintenance of the histone variant H3.3 at active regulatory elements, such as super-enhancers, and this function is required for the physiologically relevant activities of alternative chromatin remodelers.
PMID: 36153585. Journal link.
SWI/SNF (BAF) chromatin remodeling complexes regulate lineage-specific enhancer activity by promoting accessibility for diverse DNA-binding factors and chromatin regulators. Through genome-wide analysis of human endometriotic epithelial cells, we show that more than half of ARID1A binding sites are marked by the variant histone H3.3, including active regulatory elements such as super-enhancers. ARID1A knockdown leads to H3.3 depletion and gain of canonical H3.1/3.2 at ARID1A-bound active regulatory elements, and a concomitant redistribution of H3.3 toward genic elements. ARID1A interactions with the repressive chromatin remodeler CHD4 (NuRD) are associated with H3.3, and ARID1A is required for CHD4 recruitment to H3.3. ZMYND8 interacts with CHD4 to suppress a subset of ARID1A, CHD4, and ZMYND8 co-bound, H3.3+ H4K16ac+ super-enhancers near genes governing extracellular matrix, motility, adhesion, and epithelial-to-mesenchymal transition. Thus, ARID1A-containing BAF complexes are required for maintenance of the histone variant H3.3 at active regulatory elements, such as super-enhancers, and this function is required for the physiologically relevant activities of alternative chromatin remodelers.
AP‐1 Subunit JUNB Promotes Invasive Phenotypes in Endometriosis (2022, Reprod Sci)
Mike R. Wilson, Jake J. Reske, & Ronald L. Chandler.
PMID: 35616875. Journal link.
Endometriosis is a disease defined by the presence of abnormal endometrium at ectopic sites, causing pain and infertility in 10% of women. Here, we describe a role for AP-1 subunit JUNB in promoting invasive phenotypes in endometriosis. JUNB co-regulates many EMT pathway genes, such that knockdown of JUNB can reverse the invasive, mesenchymal phenotype induced by loss of ARID1A. These studies suggest a potential therapeutic mechanism targeting AP-1 in order to inhibit invasive endometriosis.
PMID: 35616875. Journal link.
Endometriosis is a disease defined by the presence of abnormal endometrium at ectopic sites, causing pain and infertility in 10% of women. Here, we describe a role for AP-1 subunit JUNB in promoting invasive phenotypes in endometriosis. JUNB co-regulates many EMT pathway genes, such that knockdown of JUNB can reverse the invasive, mesenchymal phenotype induced by loss of ARID1A. These studies suggest a potential therapeutic mechanism targeting AP-1 in order to inhibit invasive endometriosis.
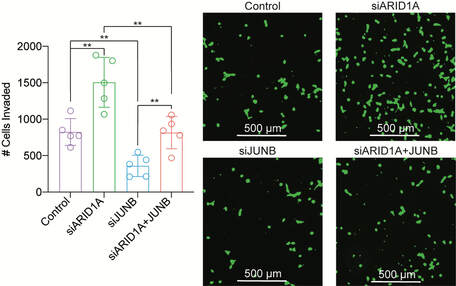
SWI/SNF Antagonism of PRC2 Mediates Estrogen-Induced Progesterone Receptor Expression (2022, Cells)
Mike R. Wilson*, Jake J. Reske*, Julie Koeman, Marie Adams, Niraj R. Joshi, Asgerally T. Fazleabas & Ronald L. Chandler. *These authors contributed equally.
PMID: 35326450. Journal link. Published as part of the Special Issue on Progesterone Receptor Signaling.
Early stage endometrial disease can often be treated with progestins, but response depends on progesterone receptor (PGR) expression. Here, we show that ARID1A loss prevents the upregulation of progesterone receptor following estrogen treatment. Additionally, we find that PGR is regulated by bivalent chromatin, harboring both H3K27me3 and H3K4me3 histone modifications. The phenotype of progesterone receptor loss can be partially rescued by inhibition of PRC2 subunit EZH2, a histone methyltransferase which often acts in an antagonistic manner to the SWI/SNF complex. Our results suggest the therapeutic utility in targeting EZH2 to restore progestin responses.
PMID: 35326450. Journal link. Published as part of the Special Issue on Progesterone Receptor Signaling.
Early stage endometrial disease can often be treated with progestins, but response depends on progesterone receptor (PGR) expression. Here, we show that ARID1A loss prevents the upregulation of progesterone receptor following estrogen treatment. Additionally, we find that PGR is regulated by bivalent chromatin, harboring both H3K27me3 and H3K4me3 histone modifications. The phenotype of progesterone receptor loss can be partially rescued by inhibition of PRC2 subunit EZH2, a histone methyltransferase which often acts in an antagonistic manner to the SWI/SNF complex. Our results suggest the therapeutic utility in targeting EZH2 to restore progestin responses.
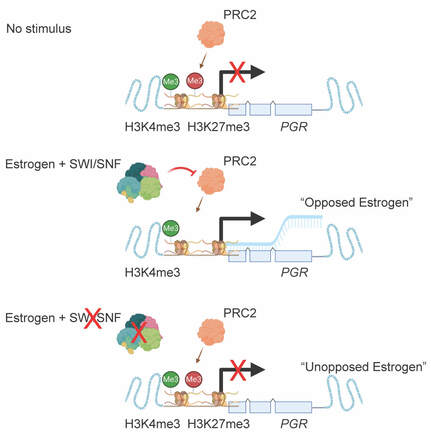
Co-existing TP53 and ARID1A mutations promote aggressive endometrial tumorigenesis (2021, PLOS Genetics)
Jake J. Reske, Mike R. Wilson, Jeanne Holladay, Rebecca A. Siwicki, Hilary Skalski, Shannon Harkins, Marie Adams, John I. Risinger, Galen Hostetter, Ken Lin, & Ronald L. Chandler.
PMID: 34941867. Journal link.
Here, we used genetically engineered mice, in vivo genomics, and public cancer patient data to understand the relationship between TP53 and ARID1A, two of the most commonly mutated genes in endometrial cancer, in the context of mutant PIK3CA. Mutations in TP53 and ARID1A change different aspects of endometrial cell health but also share some similarities. ARID1A mutations specifically promote cancer cells to invade nearby tissue, a hallmark of metastasis, associated with squamous differentiation. Mice with co-existing TP53 and ARID1A mutations developed more invasive disease. Our studies suggest that co-existing TP53 and ARID1A tumor mutations may promote invasion and metastasis.
PMID: 34941867. Journal link.
Here, we used genetically engineered mice, in vivo genomics, and public cancer patient data to understand the relationship between TP53 and ARID1A, two of the most commonly mutated genes in endometrial cancer, in the context of mutant PIK3CA. Mutations in TP53 and ARID1A change different aspects of endometrial cell health but also share some similarities. ARID1A mutations specifically promote cancer cells to invade nearby tissue, a hallmark of metastasis, associated with squamous differentiation. Mice with co-existing TP53 and ARID1A mutations developed more invasive disease. Our studies suggest that co-existing TP53 and ARID1A tumor mutations may promote invasion and metastasis.
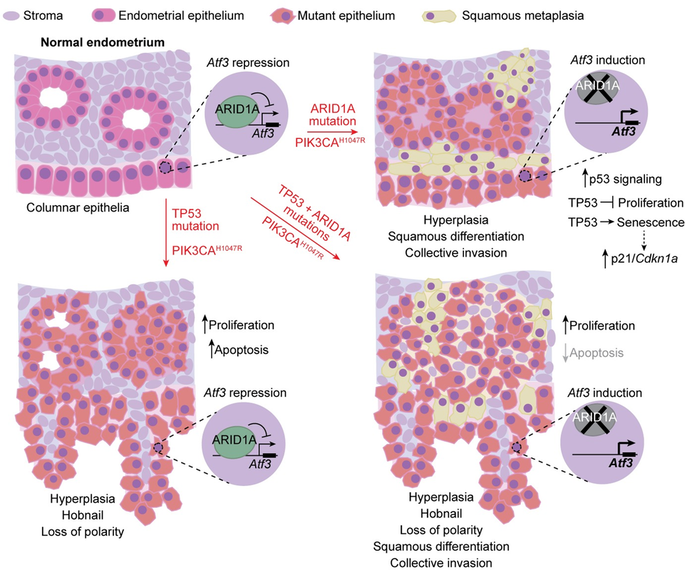
ARID1A Mutations Promote P300-Dependent Endometrial Invasion through Super-Enhancer Hyperacetylation (2020, Cell Reports)
Mike R. Wilson*, Jake J. Reske*, Jeanne Holladay, Subechhya Neupane, Julie Ngo, Nina Cuthrell, Marc Wegener, Mary Rhodes, Marie Adams, Rachael Sheridan, Galen Hostetter, Fahad T. Alotaibi, Paul J. Yong, Michael S. Anglesio, Bruce A. Lessey, Richard E. Leach, Jose M. Teixeira, Stacey A. Missmer, Asgerally T. Fazleabas, & Ronald L. Chandler. *These authors contributed equally.
PMID: 33176148. Journal link.
News coverage: MSU Today, WLNS, WILX, Genetic Engineering and Biotechnology News.
Mutations in the SWI/SNF subunit ARID1A are observed in deeply invasive endometriosis and ovarian endometriomas. Here, we show that ARID1A regulates H3K27-acetylation in the normal endometrium, such that loss of ARID1A results in H3K27ac-hyperacetylation at important genomic regions known as super-enhancers. Genetic loss or small molecule inhibition of the histone acetyltransferase P300 in ARID1A-mutant endometrium rescues super-enhancer hyperacetylation and inhibits invasive phenotypes. Specifically, hyperacetylation of the SERPINE1 (PAI-1) super-enhancer drives invasion in ARID1A-mutant endometrium. Our findings suggest that invasive endometriosis may be sensitive to super-enhancer-targeted therapeutics.
PMID: 33176148. Journal link.
News coverage: MSU Today, WLNS, WILX, Genetic Engineering and Biotechnology News.
Mutations in the SWI/SNF subunit ARID1A are observed in deeply invasive endometriosis and ovarian endometriomas. Here, we show that ARID1A regulates H3K27-acetylation in the normal endometrium, such that loss of ARID1A results in H3K27ac-hyperacetylation at important genomic regions known as super-enhancers. Genetic loss or small molecule inhibition of the histone acetyltransferase P300 in ARID1A-mutant endometrium rescues super-enhancer hyperacetylation and inhibits invasive phenotypes. Specifically, hyperacetylation of the SERPINE1 (PAI-1) super-enhancer drives invasion in ARID1A-mutant endometrium. Our findings suggest that invasive endometriosis may be sensitive to super-enhancer-targeted therapeutics.
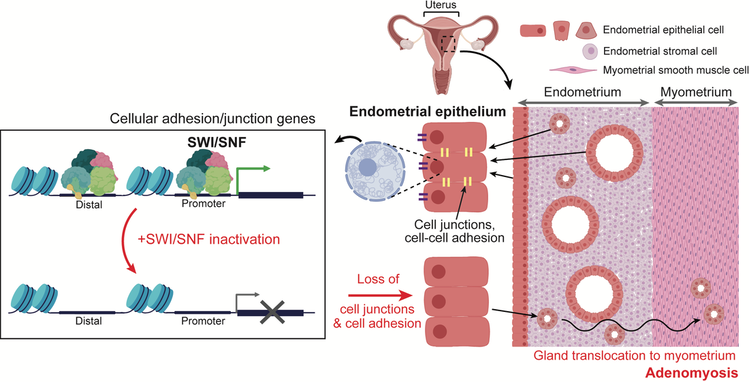
SWI/SNF inactivation in the endometrial epithelium leads to loss of epithelial integrity (2020, Human Molecular Genetics)
Jake J. Reske*, Mike R. Wilson*, Jeanne Holladay, Marc Wegener, Marie Adams, & Ronald L. Chandler. *These authors contributed equally.
PMID: 33075803. Journal link.
In endometrial cancer, ARID1A is the most frequently mutated subunit within the SWI/SNF chromatin remodeling complex, though mutations in other subunits are also observed. Here, we studied the effects of endometrial mutations in BRG1, which is one of two major catalytic subunits in the complex, and compare them to ARID1A. Through genetic models in human endometrial epithelial cells and mice, we found that BRG1 and ARID1A both normally prevent cellular invasion by promoting expression of epithelial identity genes involved in cellular adhesion and cell-cell junctions, even though they often regulate transcription in opposite ways at other genes across the genome.
PMID: 33075803. Journal link.
In endometrial cancer, ARID1A is the most frequently mutated subunit within the SWI/SNF chromatin remodeling complex, though mutations in other subunits are also observed. Here, we studied the effects of endometrial mutations in BRG1, which is one of two major catalytic subunits in the complex, and compare them to ARID1A. Through genetic models in human endometrial epithelial cells and mice, we found that BRG1 and ARID1A both normally prevent cellular invasion by promoting expression of epithelial identity genes involved in cellular adhesion and cell-cell junctions, even though they often regulate transcription in opposite ways at other genes across the genome.
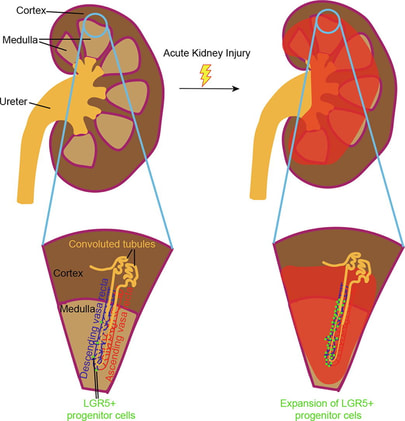
Lgr5-positive endothelial progenitor cells occupy a tumor and injury prone niche in the kidney vasa recta (2020, Stem Cell Research)
Mike R. Wilson*, Jeanne Holladay*, Rachael Sheridan, Galen Hostetter, Bree Berghuis, Carrie Graveel, Curt Essenburg, Anderson Peck, Thai H. Ho, Melissa Stanton, & Ronald L. Chandler. *These authors contributed equally.
PMID: 32464345. Journal link.
The Leucine Rich Repeat Containing G Protein-Coupled Receptor 5 (LGR5) labels tumor-prone stem cell populations in certain types of tissue. In this study, we show that ARID1A and PIK3CA mutations in LGR5-expressing cells result in renal angiosarcomas in adult mice. We further show that LGR5 labels endothelial progenitor cells within the descending vasa recta of the kidney, which give rise to renal medullary vasculature and are activated in response to ischemic kidney injury.
PMID: 32464345. Journal link.
The Leucine Rich Repeat Containing G Protein-Coupled Receptor 5 (LGR5) labels tumor-prone stem cell populations in certain types of tissue. In this study, we show that ARID1A and PIK3CA mutations in LGR5-expressing cells result in renal angiosarcomas in adult mice. We further show that LGR5 labels endothelial progenitor cells within the descending vasa recta of the kidney, which give rise to renal medullary vasculature and are activated in response to ischemic kidney injury.
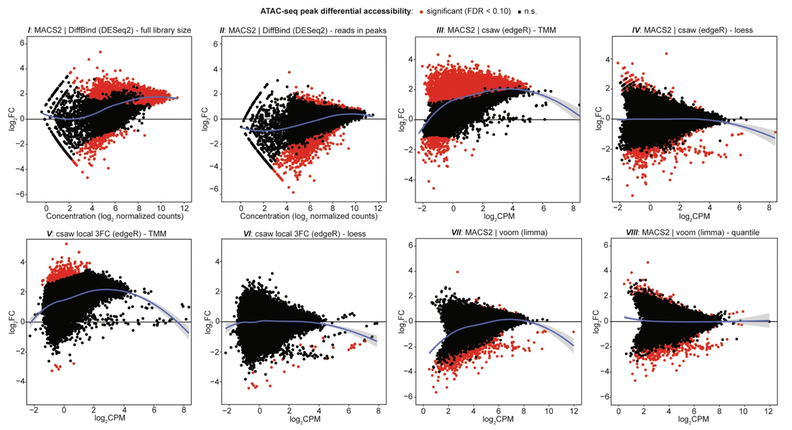
ATAC-seq normalization method can significantly affect differential accessibility analysis and interpretation (2020, Epigenetics & Chromatin)
Jake J. Reske, Mike R. Wilson, & Ronald L. Chandler.
PMID: 32321567. Journal link.
The Assay for Transposase-Accessible Chromatin (ATAC-seq) is a recently developed technique used to measure genome-wide chromatin accessibility, or where and to what extent genomic DNA is packaged within cells. This study reveals that analysis comparing ATAC-seq data between multiple cell conditions, such as with or without drug treatment, is not a straightforward procedure and can be heavily influenced by the steps used to analyze the data. Also included is a standardized bioinformatics workflow designed to help researchers appropriately analyze their own ATAC-seq data.
PMID: 32321567. Journal link.
The Assay for Transposase-Accessible Chromatin (ATAC-seq) is a recently developed technique used to measure genome-wide chromatin accessibility, or where and to what extent genomic DNA is packaged within cells. This study reveals that analysis comparing ATAC-seq data between multiple cell conditions, such as with or without drug treatment, is not a straightforward procedure and can be heavily influenced by the steps used to analyze the data. Also included is a standardized bioinformatics workflow designed to help researchers appropriately analyze their own ATAC-seq data.
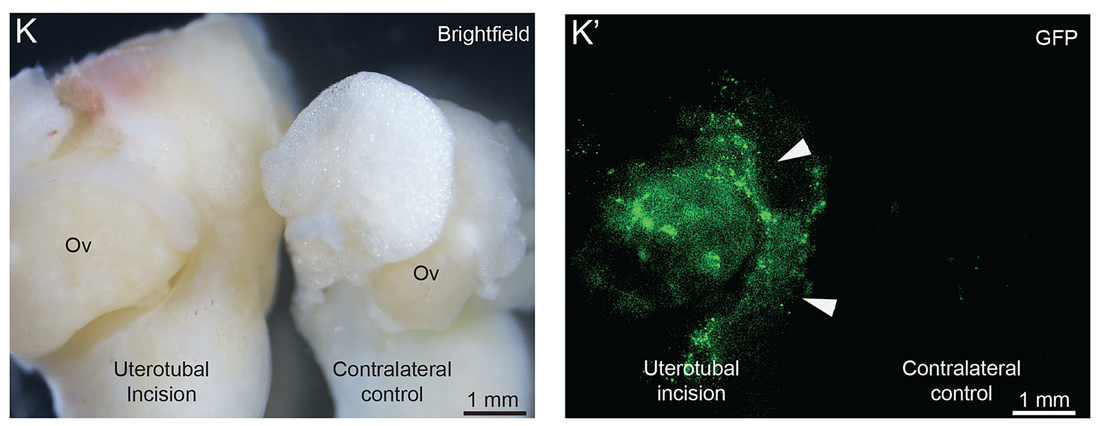
A mouse model of endometriosis mimicking the natural spread of invasive endometrium (2020, Human Reproduction)
Mike R. Wilson, Jeanne Holladay, & Ronald L. Chandler.
PMID: 31886851. Journal link.
Endometriosis is a disease in which cells lining the uterus (endometrium) grow outside of the uterus, causing pain and infertility in 5 to 10% of women. Although mouse models of endometriosis have long been established, most models rely on intraperitoneal injection of uterine fragments, steroid hormone treatments or the use of immune-compromised mice. In this study, through a combination of genetic engineering and surgical approaches, we establish a mouse model of endometriosis which mimics the natural spread of the disease from the uterus to the peritoneum, ovaries and omentum.
PMID: 31886851. Journal link.
Endometriosis is a disease in which cells lining the uterus (endometrium) grow outside of the uterus, causing pain and infertility in 5 to 10% of women. Although mouse models of endometriosis have long been established, most models rely on intraperitoneal injection of uterine fragments, steroid hormone treatments or the use of immune-compromised mice. In this study, through a combination of genetic engineering and surgical approaches, we establish a mouse model of endometriosis which mimics the natural spread of the disease from the uterus to the peritoneum, ovaries and omentum.
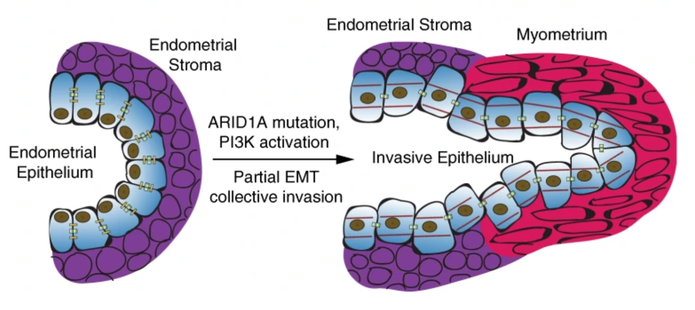
ARID1A and PI3-kinase pathway mutations in the endometrium drive epithelial transdifferentiation and collective invasion (2019, Nature Communications)
Mike R. Wilson*, Jake J. Reske*, Jeanne Holladay, Genna E. Wilber, Mary Rhodes, Julie Koeman, Marie Adams, Ben Johnson, Ren-Wei Su, Niraj R. Joshi, Amanda L. Patterson, Hui Shen, Richard E. Leach, Jose M. Teixeira, Asgerally T. Fazleabas, & Ronald L. Chandler. *These authors contributed equally.
PMID: 31391455. Journal link.
News coverage: MSU Today, The Scientist.
Mutations in the ARID1A and PIK3CA genes are common in diseases originating from the endometrial epithelium, such as endometriosis and endometrial cancer. In this study, genetically engineered mice harboring mutations in both of these genes rapidly develop vaginal bleeding and other signs of endometrial pathology, including cellular invasion. Genomic techniques are then used to study the individual and combined effects of these mutations to understand their respective contributions to endometrial disease.
PMID: 31391455. Journal link.
News coverage: MSU Today, The Scientist.
Mutations in the ARID1A and PIK3CA genes are common in diseases originating from the endometrial epithelium, such as endometriosis and endometrial cancer. In this study, genetically engineered mice harboring mutations in both of these genes rapidly develop vaginal bleeding and other signs of endometrial pathology, including cellular invasion. Genomic techniques are then used to study the individual and combined effects of these mutations to understand their respective contributions to endometrial disease.